Focus
The main focus of the organic synthesis research group is the chemical synthesis and derivatisation of organic compounds with non-trivial carbon connectivities, such as those found in polycyclic Natural Products. The design and selection of target structures is guided by a specific interest in reactivity patterns and/or a specific interest in biological phenomena. This research often follows a substrate-driven approach which relies on the use of highly modular synthetic intermediates as versatile building blocks or chemical platforms for various applications.
- The chemical synthesis research mainly involves the development of novel strategies and methods to assemble complex polycyclic scaffolds found in Nature, focusing on multiple bond forming steps such as cycloadditions and cascade reactions. [1], [2]
- The chemical derivatisation research focuses on the development of application-oriented versatile covalent ligation reactions (such as click-type reactions) to generate multiple functional derivatives from simple substrates or synthetic intermediates. [3]
- A major recent theme, which encompasses both of the above general topics, consists of research directed at the design and synthesis of highly modular synthetic building blocks that allow a rapid exploration of Natural Product-like chemical space using simple and orthogonal functional group transformations. [4]
- The concepts of versatile ligation/functionalisation reactions and modular building blocks in organic synthesis are also explored and applied in various collaborative research projects ranging from macromolecular and materials science to chemical biology. [5], [6]




Latest publications
The latest independent publications are given below, for all independent publications and collaborations, see publications

Thiol-thiol cross-clicking using bromo-ynone reagents
Thiols are used in many click reactions, and are also excellent platforms for biomolecular click or bioconjugation reactions. The direct cross-coupling of two thiols is an attractive biomimetic concept for click chemistry, but leads to statistical mixtures of homo- and heterodimers. Here, we introduce a novel class of thiol-click reagents, bromo-ynones, where the kinetic differentiation between the first and second thiol addition onto these reagents facilitates a stepwise one-pot cross-clicking of two distinct thiols in aqueous media, without the need for intermediate isolation or purification. The two thiols are linked through a single carbon atom, mimicking a disulfide bridge. We demonstrate the use of bromo-ynones in the synthesis of various cross-coupled thiols, including small molecule drugs, fluorophores, carbohydrates, peptides and proteins, including an example of a protein-protein heterodimer. The resulting adducts are robust under physiological conditions and by judicious choice of the bromo-ynone reagent, the adducts can be stable even in the presence of excess free thiols.
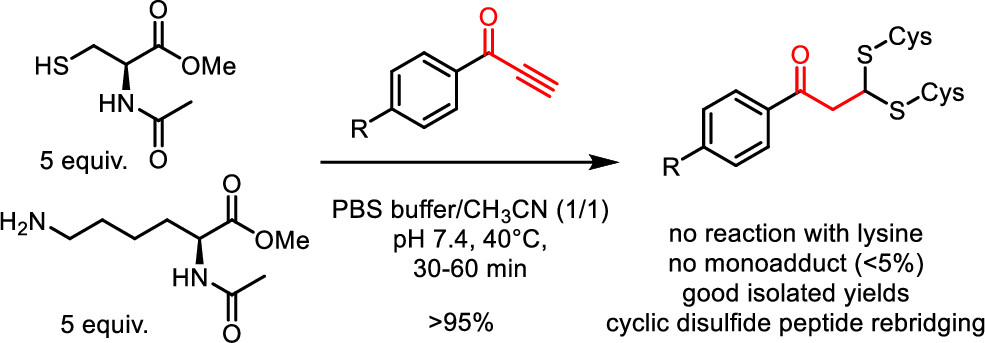
Phenylpropynones as Selective Disulfide Rebridging Bioconjugation Reagents
Simple 1-phenylpropynones undergo a selective double thia-Michael addition with thiols in buffered media, yielding an interesting dithioacetal linkage joining two thiols. The reactivity of various Michael-alkyne reagents is compared in this chemoselective, atom economical, and non-oxidative cross-linking of two thiols. The stability and chemical reactivity of the dithioacetal links are studied, and the utility of the disulfide targeting bioconjugation methodology is shown by the selective rebridging of native cyclic peptides after the reductive cleavage of their disulfide bridge.

Thermally Triggered Triazolinedione-Tyrosine Bioconjugation with Improved Chemo- and Site-Selectivity
A bioconjugation strategy is reported that allows the derivatization of tyrosine side chains through triazolinedione-based "Y-clicking". Blocked triazolinedione reagents were developed that, in contrast to classical triazolinedione reagents, can be purified before use, can be stored for a long time, and allow functionalization with a wider range of cargoes and labels. These reagents are bench-stable at room temperature but steadily release highly reactive triazolinediones upon heating to 40 °C in buffered media at physiological pH, showing a sharp temperature response over the 0 to 40 °C range. This conceptually interesting strategy, which is complementary to existing photo- or electrochemical bioorthogonal bond-forming methods, not only avoids the classical synthesis and handling difficulties of these highly reactive click-like reagents but also markedly improves the selectivity profile of the tyrosine conjugation reaction itself. It avoids oxidative damage and "off-target" tryptophan labeling, and it even improves site-selectivity in discriminating between different tyrosine side chains on the same protein or different polypeptide chains. In this research article, we describe the stepwise development of these reagents, from their short and modular synthesis to small-molecule model bioconjugation studies and proof-of-principle bioorthogonal chemistry on peptides and proteins.

Dearomative (3 + 2) Cycloaddition of Indoles for the Stereoselective Assembly of Fully Functionalized Cyclopentanoids
The gold(I)-catalyzed dearomative cyclopentannulation of various indoles with 2-ethynyl-1,3-dithiolane is reported. The method generates three new stereocenters with excellent control of relative stereochemistry and is tolerant of diverse functionalization and substitution patterns on the indoles. The obtained cyclopentane-fused indolines allow for interesting subsequent synthetic manipulations, giving rise to fully substituted cyclopentanes with control of the relative stereochemistry of all five stereocenters. The reported reaction illustrates and elucidates a mechanistic dichotomy underlying gold(I)-catalyzed reactions of 2-ethynyl-1,3-dithiolane.

